





24小時服務熱線:
400-1846-454




激光水平儀出口美國市場需要完成FDA注冊,這并非傳統意義上的"認證",而是美國食品藥品監督管理局(FDA)對
激光輻射類產品實施的市場準入管制。以下是關鍵要點:
法規依據
激光水平儀受FDA下屬的設備和放射健康中心(CDRH)監管,核心法規為21 CFR 1040.10(激光產品性能標準)和
21 CFR 1040.11(特定激光產品要求)。這些法規以法律形式強制要求,違反將面臨法律后果,其效力高于普通行業
標準。
激光安全等級分類
FDA將激光產品按危害程度分為I至IV類(對應IEC 60825-1的1至4級):
I類:無生物性危害,激光系統在暴露時互鎖
II類:輸出功率≤1mW,眨眼反射可防止眼部損傷
IIIa類:輸出功率1-5mW,可見光范圍內,直視可能造成損傷
IIIb類:輸出功率5-500mW,對皮膚和眼睛有急性危害
IV類:輸出功率>500mW,可造成嚴重眼睛損傷、灼傷皮膚甚至引發火災
大多數商用激光水平儀屬于II類或IIIa類,需滿足相應安全要求。
核心合規要求
產品標簽:必須包含激光輻射警告標志、輸出功率、波長信息、符合FDA規定的安全說明,且標簽需使用耐久材料
制作。
安全功能:根據等級配備保護外殼、安全聯鎖、鑰匙控制、光束衰減器、遠程聯鎖連接器等工程控制措施。
技術文件:需提供激光安全測試報告(依據21 CFR 1040.10或IEC 60825-1+差異條款)、產品結構圖、激光發射器
規格、英文用戶手冊(含安全使用指南)。
注冊流程
確定激光等級:通過實驗室測試確定產品所屬安全等級
準備技術文件:包括測試報告、產品規格、標簽樣本、質量控制程序文件等
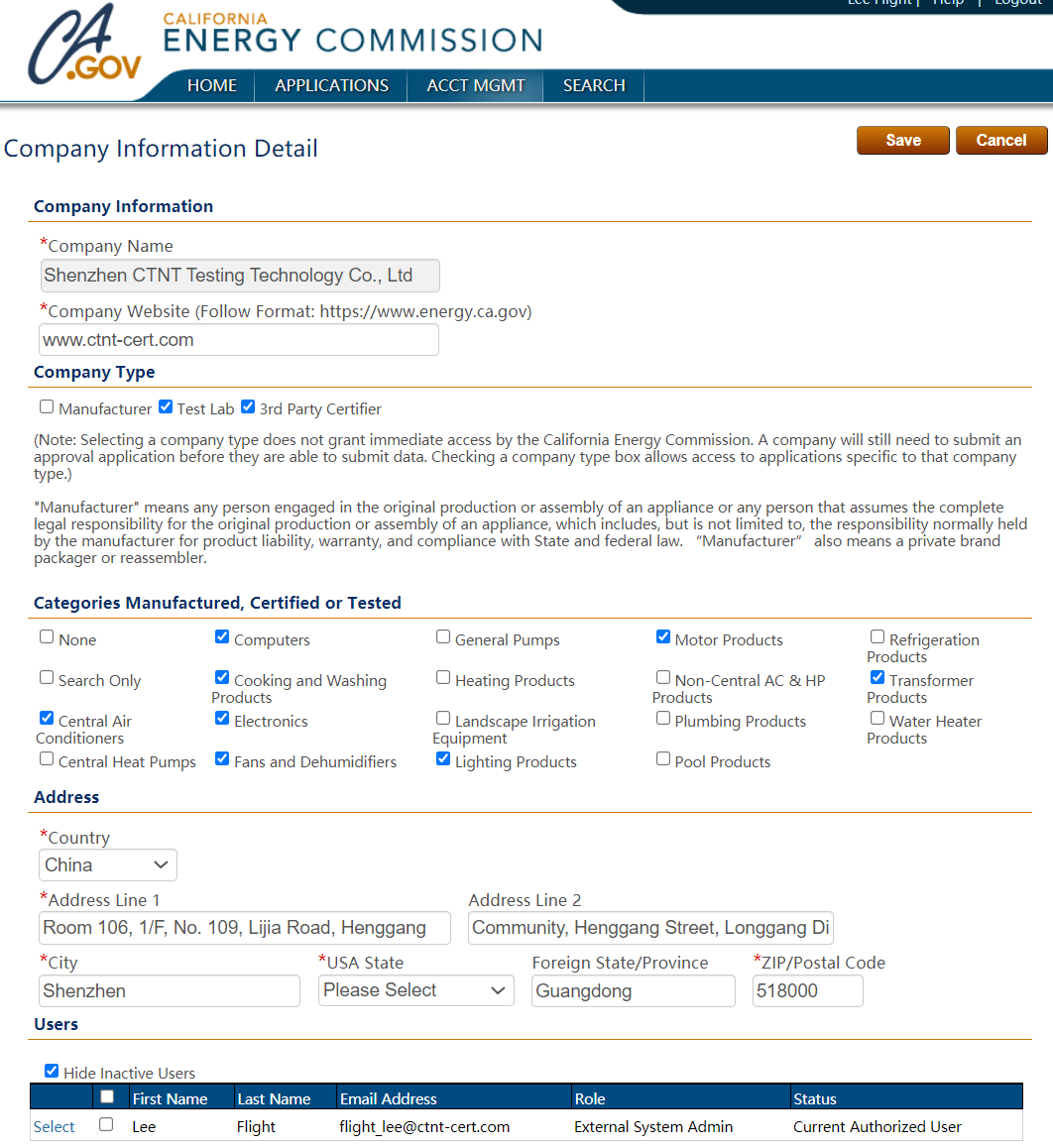
企業注冊與產品列名:制造商需在FDA官網完成企業注冊(Establishment Registration),提交產品列名
(Device Listing)
提交產品報告:使用FDA 3632表格提交激光產品報告(Laser Product Report),獲取FDA準入號
(Accession Number)
年度報告:每年9月1日前提交年度報告,包含質量控制程序描述及輻射安全測試結果
整個流程在資料齊全情況下通常需要約2周完成注冊認證環節。
測試標準選擇
制造商可選擇以下兩種路徑之一:
路徑一:直接按照21 CFR 1040.10進行檢測,這是最直接的合規方式。
路徑二:如已按IEC 60825-1檢測,可依據FDA第50號公告(Laser Notice No. 50)采用等效要求替代部分條款,但
需注意部分條款不可替代,包括認證標簽、識別標識、差異申請、可移除激光系統、手動復位機制、采購和維修信息、
產品修改以及測量/水準/對準激光產品的特定要求。
關鍵注意事項
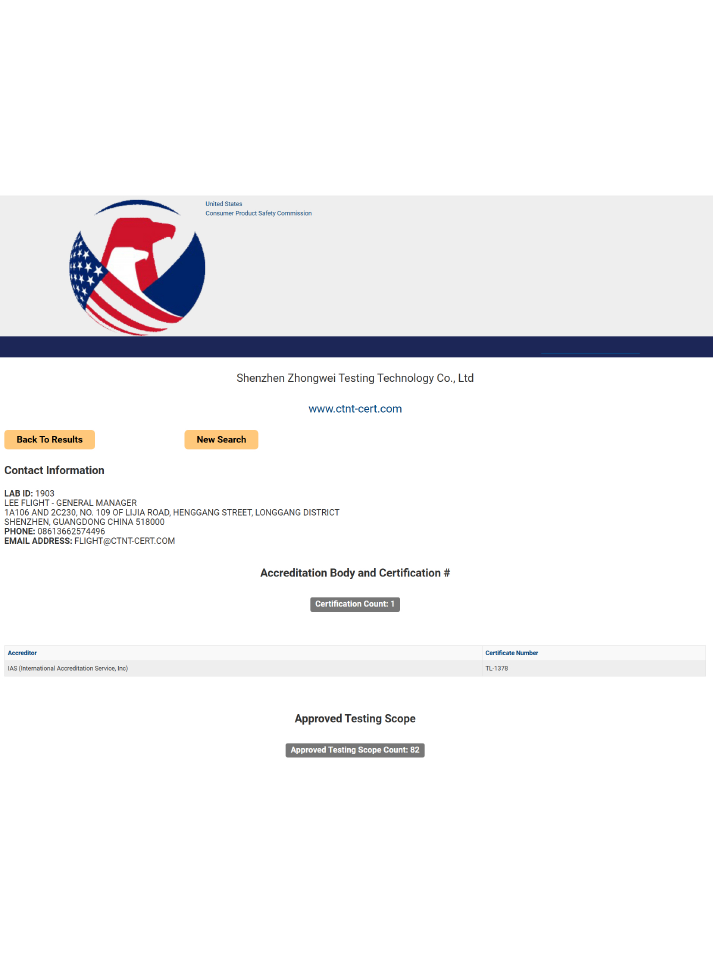
無實體證書:FDA不會頒發實體認證證書,僅保留注冊信息并出具注冊號,但產品必須帶有符合規定的認證標簽。
海關清關:獲得Accession Number是順利清關并在美國市場銷售的必要條件,未注冊產品可能被海關扣留或召回。
設計變更管理:通過認證后,重大設計變更需重新申報,FDA實行年度隨機抽檢制度。
多型號要求:各型號產品需分別提交測試數據,不能以單一型號測試結果套用全系列產品






返回頂部

電話:400-1846-454

地址:深圳市龍崗區橫崗街道橫崗社區力嘉路109號
 當前位置:
當前位置: 





 網站備案:
網站備案: 公安網備案:
公安網備案:

