





24小時服務熱線:
400-1846-454




如今跨境電商賽道競爭加劇,激光設備作為高關注度品類,在美國亞馬遜平臺銷售必須嚴守FDA 強制合規底線。激光產品屬于輻射類電子設備,未完成FDA認證與備案,輕則Listing審核駁回、產品下架,重則海關扣貨、面臨高額處罰。本文結合 2026 年最新法規與亞馬遜平臺規則,為激光設備賣家梳理完整FDA認證流程、分級要求、文件清單及避坑要點,助力產品合規出海、穩定運營。
")
一、激光設備FDA認證核心法規依據
美國對激光產品的監管由FDA 下屬 CDRH(輻射健康中心) 執行,核心法規為21 CFR 1040.10(通用激光產品性能標準)與21 CFR 1040.11(專用激光產品補充要求),覆蓋所有在美國流通的激光設備,無論產地與用途,均需滿足此標準。
亞馬遜作為平臺方,嚴格對接 FDA 監管要求,無 FDA 合規文件一律禁止上架,且會定期復核合規資質,違規產品直接下架清退。
二、FDA 激光設備分級標準與合規要求
FDA按激光輻射危害程度分為4大類,等級越高監管越嚴,直接決定測試項目、標簽規范與文件復雜度:
ClassⅠ類:功率<0.39mW,正常使用無安全風險,如激光內置模塊、低功率掃描設備。需滿足基礎標識、產品報告要求,無需復雜防護裝置。
Class Ⅱ 類:0.39–1mW 可見光激光,如激光筆、條碼掃描儀。短暫直視可通過眨眼反射防護,需標注CAUTION警示語與等級信息。
Class Ⅲa 類:1–5mW 中功率激光,直視易傷眼,必須張貼清晰警告標簽、配備安全說明,禁止無防護銷售。
Class Ⅲb/Ⅳ 類:功率>5mW 高功率激光,可致眼、皮膚永久損傷,甚至引發火災。需安全聯鎖、鑰匙開關、光束終止器等防護裝置,文件與測試要求最嚴格。
數據顯示,亞馬遜平臺超8成激光產品審核失敗,源于分級錯誤、認證缺失,精準分級是合規第一步。
三、2026年激光設備FDA認證核心流程(亞馬遜上架必備)
1. 產品合規檢測(ISO 17025實驗室出具報告)
認證前提是通過權威檢測,核心項目包括:
激光輸出功率、波長、能量密度精準測量
輻射分布、發散角、脈沖特性評估
安全裝置(聯鎖、鑰匙開關、報警)功能驗證
標簽、說明書合規性審核
測試報告需為近 12 個月內整機檢測報告,組件合格報告無效,亞馬遜與FDA均不認可。
2.FDA激光產品報告(LRP)備案
所有激光設備必須向 FDA 提交Laser Product Report,核心材料:
產品技術規格、結構原理說明
制造商信息、符合性聲明
生產質量控制程序文件
產品標簽樣張、完整使用說明書
第三方實驗室檢測報告
備案后獲取 FDA 產品登記號,這是亞馬遜審核的核心憑證。
3. 標簽與說明書強制規范(缺一不可)
產品外殼需永久粘貼合規標簽,包含:
對應等級警示詞(DANGER/CAUTION)
激光等級標識(如 CLASS Ⅱ LASER PRODUCT)
輸出功率、波長參數
制造商名稱、地址
合規聲明(Complies with 21 CFR 1040.10)
說明書需包含安全操作、維護流程、潛在危害提示,高等級產品需增加專業培訓說明。
4. 質量控制與記錄留存
制造商需建立全流程質控體系,保留生產檢測記錄,至少留存至停產后5年,應對FDA隨機抽查。
四、亞馬遜美國站激光產品上架文件清單
創建Listing前,需在賣家中心提交全套合規文件,缺一不可:
FDA激光產品報告備案確認頁(含產品登記號)
ISO 17025認可實驗室出具的檢測報告
產品標簽、說明書高清樣張
制造商符合性聲明(DCL)
企業 FDA 注冊信息(FEI編碼)
數據顯示,文件齊全的賣家上架通過率超98%,缺失文件首次駁回率超80%。
五、2026年FDA認證高頻誤區(避坑必看)
CE 認證≠FDA 合規
歐盟CE認證與美國FDA體系完全獨立,僅持CE認證上架,亞馬遜直接暫停 Listing,2022年超200個鏈接因此下架。
自行分級風險極高
非專業判斷易導致分級偏低,后續被FDA核查將面臨召回、罰款,建議委托第三方機構精準分級。
產品變更未更新認證
功率、結構、配件改動后,需重新檢測并更新FDA報告,約30%合規問題源于此。
忽略州級附加規則
加州等州對激光筆、寵物激光玩具等有額外限制,需同步滿足聯邦與州級要求。
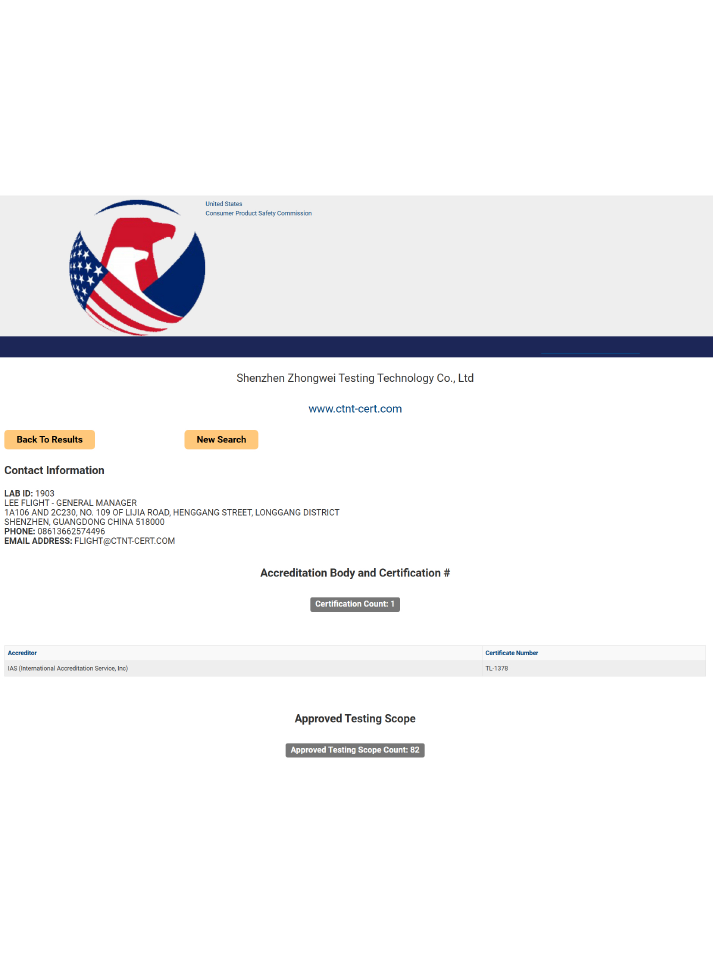
六、中為檢驗技術——專業的第三方激光檢測機構
一次過審:精通FDA與亞馬遜規則,避免分類錯誤、材料缺失,大幅提升審核效率。
縮短周期:專業團隊代辦備案、測試、溝通,認證周期平均縮短40%,加速上架。
風險兜底:提前排查設計缺陷,提供整改方案,規避召回、處罰、下架風險。
長期合規:提供產品變更評估、年度報告、合規年審,保障長期穩定銷售。
如您有激光產品檢測方面的需求,歡迎咨詢深圳中為檢驗業務電話:18038017984(V信同號)






返回頂部

電話:400-1846-454

地址:深圳市龍崗區橫崗街道橫崗社區力嘉路109號
 當前位置:
當前位置: 





 網站備案:
網站備案: 公安網備案:
公安網備案:

